
Poor adhesion between an organic polymer matrix and an inorganic filler or substrate is not a materials science abstraction — it shows up as delamination in rubber-coated rolls, premature cracking in glass-fiber composites, and moisture-induced bond failures in sealed electronics assemblies. Each of those failures carries a real cost: scrapped production runs, warranty returns, or unplanned line stoppages to re-coat or re-cure. The root cause is almost always the same chemical incompatibility at the interface, and silane coupling agents exist specifically to bridge it. Understanding their chemical structures is not academic; it determines which product you specify, how you formulate it, and whether the coupling chemistry actually completes at the temperatures and pH conditions on your process line.
A silane coupling agent is an organosilicon molecule carrying two chemically distinct ends on the same backbone: a hydrolyzable inorganic-reactive group — typically three alkoxy groups bonded to silicon — and an organofunctional group, such as an amine, epoxy, methacrylate, or sulfide, tethered to silicon through a short carbon spacer. The inorganic end bonds to hydroxylated mineral surfaces or glass through silanol condensation; the organic end reacts with or is compatible with the polymer matrix. That dual reactivity in a single small molecule is what makes them effective as adhesion promoters, surface modifiers, and crosslinking agents across a wide range of industrial applications.
What makes the chemistry genuinely interesting — and practically consequential — is that neither end operates independently of the other. The spacer length, the choice of alkoxy leaving group, and the nature of the organofunctional group all interact to govern hydrolysis rate, condensation behavior, and final bond geometry. A three-carbon propyl bridge between silicon and an amine group behaves very differently from a one-carbon or six-carbon version, even though both are technically “aminosilanes.” Getting into the structural details reveals why certain silanes dominate commercial procurement and why others fail quietly in production without ever triggering an obvious alarm.
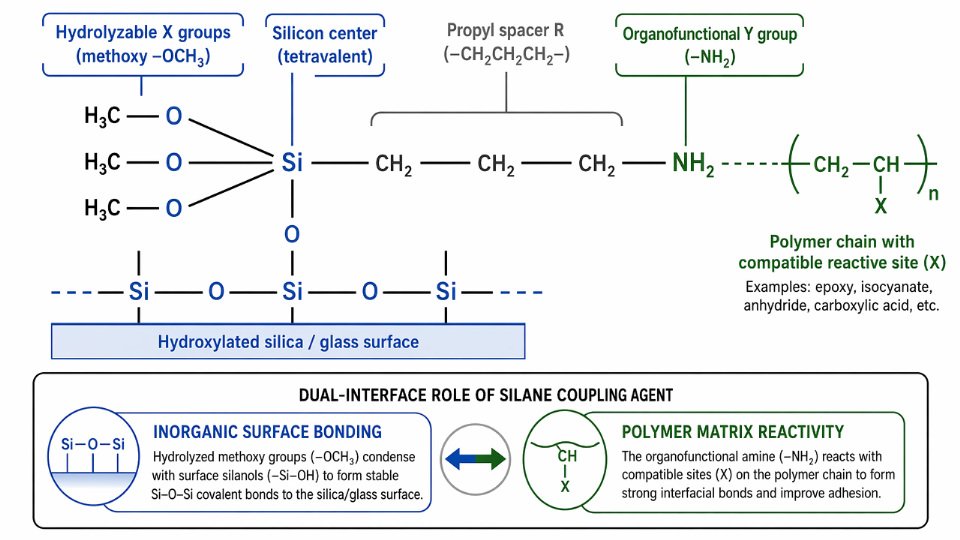
The General Molecular Formula X₃Si–R–Y: Parsing Every Atom and Bond
Silicon sits at the center of every silane coupling agent molecule, and that placement is not arbitrary. Silicon is tetravalent — it forms exactly four covalent bonds simultaneously, no more, no fewer. In the canonical trialkoxysilane architecture, three of those four bonds go to hydrolyzable X groups, and the fourth anchors the organic arm R–Y. That single silicon atom is therefore doing two jobs at once: presenting an inorganic-compatible face toward mineral surfaces, metals, and glass, while holding out an organic-compatible arm toward polymer matrices. Everything else in the molecule is subordinate to that dual-interface role.
The X Group: Hydrolyzable Anchor to Inorganic Surfaces
The X groups are what allow the silane to bond to hydroxyl-bearing surfaces. In water — or simply in the presence of atmospheric moisture — X groups hydrolyze to silanols (Si–OH), which then condense with surface hydroxyls to form stable Si–O–Si bonds. The four commercial X group types each carry practical trade-offs.
Methoxy (–OCH₃) is the workhorse for fast-coupling applications. Under dilute aqueous conditions at pH 4–5 and 25°C, trimethoxysilanes hydrolyze in roughly 10–30 seconds. The by-product is methanol — low molecular weight, volatile, and regulated in enclosed processing environments. Ethoxysilanes hydrolyze 3–5 times more slowly than their methoxy counterparts under equivalent conditions, releasing ethanol instead. That slower rate is actually an advantage in formulations where pot life matters, such as surface treatment baths that must remain stable for hours. Acetoxysilanes (–OOCCH₃) hydrolyze rapidly but release acetic acid, which is corrosive to certain metals and copper-containing electronics substrates — a real constraint in PCB laminate applications. Chlorosilanes hydrolyze fastest of all and generate HCl, making them nearly exclusive to controlled reactor synthesis rather than end-use application.
Methoxy silanes hydrolyze approximately 3–5 times faster than ethoxy silanes under equivalent pH and temperature conditions.True
Hydrolysis rate depends on the leaving-group ability of the alkoxide; methanol is a better leaving group than ethanol under mild acidic conditions, which is consistent with published kinetic data for trialkoxysilane hydrolysis.
Shelf-life follows the same ranking in reverse: chlorosilanes and methoxysilanes need rigorous moisture exclusion during storage, while ethoxysilanes tolerate moderate humidity with proper container sealing.
The R Spacer: More Than a Simple Bridge
The R group connects silicon to the functional end of the molecule, and its length and flexibility are not trivial choices. A three-carbon propyl chain (–CH₂CH₂CH₂–) dominates commercial production, accounting for the majority of aminosilane and most other functionalized silane architectures sold today. That propyl bridge is long enough to provide conformational freedom — the Y group can orient toward incoming polymer chains rather than being sterically pinned against the silica surface — yet short enough to keep molecular weight low and surface silanol density high per unit area.
Shorter bridges reduce that flexibility and can cause steric crowding at the interface, limiting how efficiently silanols pack and condense during bonding. Longer chains increase molecular weight, dilute the functional group concentration, and can introduce unwanted mobility that weakens the interface under thermal cycling.
The Y Group: Where Polymer Chemistry Enters
The Y group is the organofunctional terminus, and selecting it correctly is the entire basis of coupling agent specification. Amino (–NH₂), epoxy (glycidoxy), vinyl, methacryloxy, mercapto (–SH), and isocyanato groups each react with a specific set of polymer chemistries — thermosets, elastomers, thermoplastics, and UV-cure systems all call for different Y groups. This catalogue gets detailed treatment in subsequent sections, but the structural principle holds here: Y must be chemically reactive toward the matrix and chemically inert toward the inorganic substrate, the inverse of what X does.
Bis-Silanes and Monoalkoxysilanes: Beyond the Standard Template
Not every commercial silane fits the simple X₃Si–R–Y template. Bis-silanes carry two trialkoxysilyl ends bridged by a single organic chain — TESPT (bis[3-(triethoxysilyl)propyl] tetrasulfide), used in silica-reinforced tire compounds, is the highest-volume example. The second silane end increases cross-link density within the silica–rubber interphase, which measurably reduces rolling resistance compared to mono-functional analogs. The trade-off is higher cost and more demanding mixing temperature control during compounding; if mixing temperature exceeds roughly 150°C prematurely, the sulfide bridge can scorch the rubber before adequate silane coverage is achieved. Monoalkoxysilanes sacrifice condensation cross-linking entirely but gain water resistance in cured films, useful in moisture-sensitive coatings where residual silanol groups would be a liability rather than an asset.
The Hydrolyzable Inorganic End: Silanol Formation, Condensation, and Substrate Bonding Mechanisms
The inorganic end of a silane coupling agent does not bond to a substrate in the form you purchase it. That point alone corrects one of the most persistent misconceptions on plant floors and in formulation labs. The alkoxy groups — methoxy or ethoxy, most commonly — must first be converted to silanols before any meaningful surface chemistry can begin. Understanding the three-step sequence from hydrolysis through condensation to covalent surface attachment is not academic; it directly determines whether you get a durable bonded interface or a layer that washes off under humidity cycling.
Step 1: Hydrolysis and the Formation of the True Reactive Species
The reaction X₃Si–R–Y + 3H₂O → (HO)₃Si–R–Y + 3HX releases the alkoxy leaving groups as their parent alcohols (methanol from trimethoxysilanes, ethanol from triethoxysilanes) and produces the trisilanol intermediate. This reaction is both acid- and base-catalyzed. In industrial surface treatment practice, a dilute acetic acid solution at pH 4–5 is the standard conditioning medium, and for good reason: trialkoxysilanes hydrolyze within roughly 10–90 seconds under those conditions at 25°C in dilute aqueous solution, with the exact rate depending on the alkoxy substituent (methoxy hydrolyzes faster than ethoxy), the organic spacer group R, and temperature. At neutral pH, hydrolysis can take minutes to hours — far too slow for continuous coating or compounding lines.
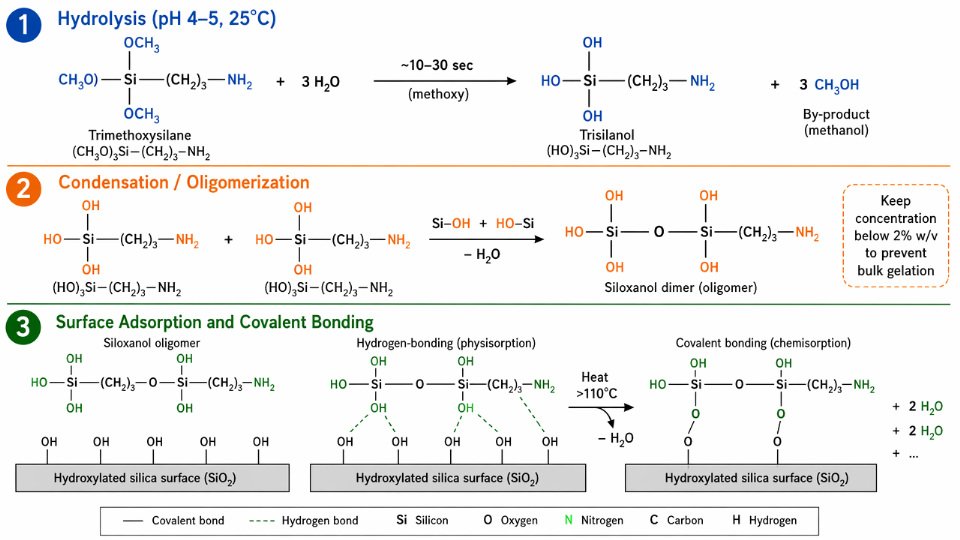
The silanol (Si–OH) is the true reactive species. Treating the unhydrolyzed alkoxysilane as an adhesion promoter and adding it directly to an aqueous system without acidification is a common formulation error that results in inconsistent coupling efficiency and batch-to-batch variability in adhesion strength.
Step 2: Condensation, Oligomerization, and Concentration Control
Silanols are unstable relative to siloxanes. The condensation reaction Si–OH + HO–Si → Si–O–Si + H₂O proceeds at measurable rates across a wide pH range — above pH 3 and below pH 10 — with the hydrolyzed intermediate showing maximum stability near pH 4–5. Left at higher concentrations or at neutral-to-basic pH, silanols rapidly condense with each other, forming dimers, trimers, and eventually oligomeric siloxanol networks that can gel out of solution entirely before reaching the substrate surface.
This is why working solutions are kept below approximately 2% w/v. Above that threshold, bulk gel formation accelerates and the silane is effectively consumed in solution rather than deposited as a functional surface film. The consequence is measurable: coupling efficiency drops, surface coverage becomes patchy, and filler dispersion in rubber or polymer composites suffers directly. For high-throughput glass fiber sizing operations, maintaining both concentration and pH within specification is a process control requirement, not a recommendation.
Silane coupling agents bond directly to surfaces through their alkoxy groups without requiring hydrolysis.False
Alkoxy groups (–OCH₃, –OC₂H₅) are not themselves reactive toward hydroxylated surfaces. Hydrolysis to silanols (Si–OH) is a prerequisite for surface condensation and covalent bonding. Skipping or inadequately completing this step produces weak physisorbed interfaces rather than covalent Si–O–substrate bonds.
Step 3: Adsorption, Covalent Bonding, and the Multilayer Deposition Model
Once siloxanol oligomers reach the substrate surface, the initial interaction is hydrogen bonding — silanol groups aligning with surface hydroxyls on silica or glass (Si–OH surface sites) or with metal hydroxide groups on aluminum and iron oxide surfaces (Al–OH, Fe–OH). This hydrogen-bonded precursor layer is mobile and reversible at room temperature.
Drying and curing above approximately 110°C drives condensation of these hydrogen bonds into covalent Si–O–substrate linkages, releasing water and locking the interface. The result is not a single molecular monolayer but typically 1–3 covalent monolayers at the substrate surface, with additional physisorbed siloxane layers stacked above. This multilayer structure matters because bond durability in humid or aqueous service environments depends critically on the covalent-to-physisorbed ratio. Physisorbed layers are removable by water; covalent layers are not.
Residual Silanols as Weak Points and the Cure Schedule Imperative
Any Si–OH group that does not condense — either with a neighboring silanol or with the substrate surface — remains in the deposited film as a hydrophilic site. These residual silanols are hydrolytic reversal points: under prolonged moisture exposure, the Si–O–Si bonds formed during cure can rehydrolyze back to silanols, degrading the interface over time. A film with a high fraction of residual silanols will show accelerated adhesion loss in boiling water tests or salt-fog exposure.
This motivates cure schedules rather than simple air drying. Insufficient cure temperature leaves the covalent network underdeveloped. Excessive temperature or time can thermally degrade the organic functional group Y at the other end of the molecule — a separate constraint that forces cure windows to be defined for each specific silane chemistry and substrate combination.
Amino Functional Groups (–NH2 and –NH–): Structures, Reactivity, and Applications in Epoxy and Polyurethane Systems
Amino-functional silanes are the single largest category in most silane coupling agent portfolios, and for good reason. The nitrogen atom’s lone electron pair gives these molecules a reactivity profile that fits cleanly into the cure chemistry of epoxy resins, polyurethanes, and several adhesive systems. Understanding the structural differences between primary amine, secondary amine, and diamine architectures directly determines whether a formulator gets a fast-cure, high-adhesion interphase or a brittle, under-crosslinked failure zone.
Primary Aminosilanes: APTES as the Structural Benchmark
3-Aminopropyltriethoxysilane — APTES, H2N–C3H6–Si(OEt)3 — is the reference molecule for this class. The three-carbon propyl spacer connecting the silicon center to the terminal –NH2 accounts for that 60–70% share of aminosilane commercial volume mentioned in the broader architecture discussion; the chain is long enough to provide conformational flexibility at the interphase but short enough to avoid steric shielding of the amine.
The –NH2 group is a primary amine. It carries two labile hydrogen atoms and a high-electron-density nitrogen, making it an effective nucleophile toward epoxide rings, isocyanates, acid anhydrides, and activated esters. In an epoxy composite system, the ring-opening reaction between –NH2 and an epoxide is exothermic and proceeds without catalyst at temperatures between roughly 60–120°C, depending on epoxide equivalent weight and stoichiometry. Each –NH2 can theoretically react with two epoxide groups, which is why amine hydrogen equivalent weight matters when calculating stoichiometry for fiber sizings or adhesive primers.
Secondary Amine and Diamine Architectures: DAMO and Cross-Link Density
N-(2-Aminoethyl)-3-aminopropyltrimethoxysilane, widely known as DAMO, packs both a primary amine at the chain terminus and a secondary amine (–NH–) along the ethylene bridge within a single molecule. The secondary amine’s single labile hydrogen reacts more slowly with epoxides than the primary amine — rate differences of roughly 3–10× are typical, depending on temperature and solvent polarity — but its contribution to cross-link density is real and useful.
Practically, DAMO raises the total amine hydrogen content available at the fiber–matrix interphase. In a glass-fiber-reinforced epoxy laminate, this translates to a denser covalent network within the 5–20 nm interphase zone. The consequence for interlaminar shear strength (ILSS) is measurable: APTES treatment on E-glass typically raises ILSS by 30–50% over unsized fiber, and DAMO can push that increment further in systems where cross-link density at the interface is the limiting factor rather than bulk matrix toughness.
APTES treatment of E-glass fiber raises interlaminar shear strength by 30–50% compared to unsized fiber in epoxy composite systemsTrue
This range is consistent with published composite interphase research and reflects variation due to fiber surface hydroxyl density, silane application method (aqueous vs. anhydrous), cure schedule, and epoxy system used. Results outside this range occur with very high or very low silane loading.
Cure kinetics change with DAMO. Because the secondary amine reacts slower, gel time extends relative to an equivalent loading of APTES. In a production prepreg or pultrusion operation, that shift in pot life needs to be factored into line speed and oven dwell time — using DAMO interchangeably with APTES without adjusting cure parameters is a common source of under-cure defects near the fiber bundle interior.
Intramolecular Hydrogen Bonding After Hydrolysis
Once the trialkoxysilane end hydrolyzes to silanol, the –NH2 group forms intramolecular hydrogen bonds with adjacent Si–OH oxygens. This is structurally stabilizing — it helps explain why aminosilane hydrolysates are somewhat less prone to rapid self-condensation than, say, glycidoxysilanes — but it also partially ties up the amine lone pair. If the application solution sits too long or dries at too low a temperature, those H-bonds persist at the surface and reduce the amine’s nucleophilic availability toward incoming epoxide groups.
Operational warning: aminosilane treating baths above pH 8 accelerate silanol self-condensation rapidly. Keep aqueous aminosilane solutions at pH 4–5 and use within 4–8 hours of preparation to maintain both hydrolytic stability and surface amine reactivity.
Corrosion Inhibition: Lone-Pair Coordination to Metal Oxides
On metal substrates — steel, aluminum, and their native oxides — amino silanes provide a dual-anchor mechanism. The silanol end forms Si–O–metal covalent bonds at surface hydroxyl sites, as with any coupling agent. The –NH2 group adds a second interaction: lone-pair donation from nitrogen to Lewis acid surface sites such as Fe³⁺ and Al³⁺. This coordinate bond is not as thermodynamically stable as a covalent Si–O–metal linkage, but it provides supplementary adhesion under humid or mechanically stressed conditions where some Si–O bonds have already hydrolyzed.
In polyurethane adhesive systems bonded to aluminum, this dual anchoring is part of why aminosilane primers outperform simple silanol-based primers under salt-spray testing — the nitrogen coordination gives a redundant contact mechanism while the Si–O–Si network carries the primary structural load.
Epoxy and Glycidoxy Functional Groups: Three-Membered Ring Strain and Ring-Opening Chemistry
3-Glycidoxypropyltrimethoxysilane — almost universally abbreviated GPTMS — is structurally distinct from amino or mercapto silanes because its organic reactive end is a ring, not an open-chain functional group. That ring, the three-membered oxirane, is what makes this the workhorse of the silane coupling agent world.
The Oxiranylmethoxy Architecture of GPTMS
Draw GPTMS from left to right and you see three zones: the trimethoxysilyl anchor (Si(OCH₃)₃), a three-carbon propyl spacer, an ether oxygen, and then the glycidyl terminus — a CH₂ group bonded to an oxirane ring. The ether oxygen between the spacer and the glycidyl group matters because it introduces conformational flexibility while also being the weakest link chemically (more on that below). The oxirane itself is a symmetric, nearly equilateral triangle of two carbons and one oxygen, with C–O bond angles forced to roughly 60° versus the relaxed ~111° preferred by sp³ oxygen. That geometric distortion stores approximately 114 kJ/mol of ring strain energy. Practically, this means the ring does not need a strong nucleophile or extreme conditions to open — it is, by thermodynamic definition, waiting to react.
Ring-Opening with Amines: The Core Industrial Reaction
When a primary amine (R–NH₂) attacks the epoxide, it opens the ring at the less-hindered carbon and produces a β-hydroxyamine: the nitrogen forms a new C–N bond and the oxygen picks up a proton to become a hydroxyl group. The reaction is exothermic, essentially irreversible under processing conditions, and proceeds without a catalyst at temperatures between 50–120°C depending on amine type, stoichiometry, and system viscosity.
This is precisely why GPTMS dominates at the interface between amine-cured epoxy matrices and inorganic substrates — glass fiber, aluminum, mineral fillers. The silane hydrolyzes and bonds to the inorganic surface through siloxane linkages; the glycidoxy end coreacts with the amine hardener in the bulk resin. You get a covalent bridge across what would otherwise be a mechanically weak, hydrolytically vulnerable interface. Omit the silane or use the wrong one and the failure mode shifts from cohesive (acceptable) to interfacial (catastrophic), typically showing up as delamination after humidity cycling or thermal shock.
Versatility Across Nucleophile Types
Thiols open epoxides fast — seconds to a few minutes with mild base catalysis at room temperature. This kinetic advantage makes GPTMS genuinely useful in UV-curable hybrid systems and rapid moisture-cure formulations where pot life is measured in minutes, not hours. Carboxylic acids react more slowly and typically require temperatures above 80°C, making them suitable for powder coatings and thermally activated adhesive films. Alcohols react slowest of all, usually requiring a Lewis acid catalyst, but this opens pathways in polyurethane and polyol-based matrix systems where amine chemistry would cause side reactions.
GPTMS ring-opening with thiols under base catalysis proceeds to high conversion at room temperature within minutes.True
Thiol-epoxide addition is among the fastest click-type reactions in polymer chemistry; with tertiary amine or phosphine catalysis at ambient temperature, conversions above 90% within 5–10 minutes are well-documented in literature and consistent with industrial formulation practice.
Thermal and Hydrolytic Stability — One Structural Weakness to Know
The ether linkage (–C–O–C–) connecting the propyl spacer to the glycidyl group is vulnerable to acid hydrolysis at elevated temperature, particularly above 150°C in the presence of moisture or acidic fillers. In demanding environments — extended exposure to hot, humid, acidic conditions — this degradation pathway can cleave the organic functional group from the spacer before it has reacted with the matrix. Ureidosilanes and isocyanatopropylsilanes carry more hydrolytically stable organic linkages and are the technically correct choice in those conditions, even though they cost more and carry their own handling constraints.
Market Position and Why Purity Directly Affects Cure Performance
GPTMS accounts for roughly 25–35% of global silane coupling agent consumption by volume, a share that reflects the dominance of epoxy-based composite and adhesive manufacturing worldwide. The figure varies with regional industry mix — higher in markets with strong fiberglass composite or electronic laminate production, lower where polyurethane chemistry predominates.
Purity is not a marketing abstraction here. Methanol and heavier siloxane oligomers are the main byproducts in GPTMS synthesis; both dilute effective silane concentration and the oligomers can prematurely cross-link during storage, producing gel particles that block spray or sizing application equipment. SiliconChemicals produces GPTMS and its ethoxy analog at industrial scale using continuous distillation, achieving greater than 98% purity. At that level, the active epoxide content is predictable batch to batch — which matters because formulators set silane loading based on calculated reactive group equivalents, not nominal weight.
Vinyl, Methacryloxy, and Acryloxy Groups: Unsaturated Functional Groups for Radical and Addition Polymerizations
The C=C double bond is the defining feature of this silane family, but calling it simply “reactive” undersells the structural logic at work. Each variant positions that unsaturation differently relative to the silicon center, the ester linkage, and any substituents on the alpha carbon — and those differences translate directly into crosslink density, hydrolytic durability, and processing latitude on the plant floor.
Vinyltrimethoxysilane and Vinyltriethoxysilane: Direct Silicon–Vinyl Proximity
In vinyltrimethoxysilane (VTMS) and its ethoxysilane counterpart (VTES), the vinyl group (CH₂=CH–) sits directly on silicon with no intervening carbon spacer. This is structurally unusual compared to most organofunctional silanes, which carry a propyl bridge. The short Si–C(vinyl) bond places the pi system in proximity to silicon’s available d-orbitals, creating a mild electron-withdrawal effect that subtly lowers electron density on the double bond — a detail relevant when formulating radical initiation conditions, since electron-poor alkenes respond differently to radical flux than electron-rich ones.
Practically, the two biggest uses are moisture-cure silane-grafted polyethylene for medium- and high-voltage cable insulation and as a coupling agent for silica fillers in unsaturated polyester (UPE) composites. In the cable insulation route, peroxide initiates free-radical grafting of the vinylsilane onto the polyethylene backbone at melt-processing temperatures, typically 160–220 °C depending on the peroxide half-life and extruder residence time. Once the silane-grafted PE is shaped and exposed to moisture, the trimethoxysilyl groups hydrolyze and condense into siloxane (Si–O–Si) crosslinks without any press vulcanization step. You achieve crosslink densities broadly comparable to peroxide-cure at a fraction of the capital cost — no autoclave, no high-temperature mold. The tradeoff is that crosslinking rate is then humidity- and temperature-dependent rather than press-controlled, which matters for dimensional stability in thick-wall cable geometries.

3-Methacryloxypropyltrimethoxysilane: The Ester Bridge as a Design Choice
MPS (also written MAPTMS) introduces two structural elements absent from vinylsilanes: a three-carbon spacer between silicon and the ester oxygen, and a methyl substituent on the alpha carbon of the methacrylate group (CH₂=C(CH₃)–COO–). The ester linkage raises polarity, which improves wetting and adhesion to polar substrates like glass, metal oxides, and dental ceramics. The methyl group on the double bond is not decorative — it sterically reduces the propagation rate constant (k_p) in radical polymerization relative to the unsubstituted acrylate analog. That lower reactivity is often an asset: it gives formulators longer working windows in UV-cure coatings and reduces the risk of premature homopolymerization in storage.
MPS is the silane of choice in dental composite resins, where it simultaneously bonds silica filler particles covalently to the methacrylate resin matrix and participates in the crosslinked organic network formed on photocure. The same bifunctional logic applies in optical fiber coatings and display glass bonding: one molecule anchors the organic UV-cured network to the inorganic substrate, eliminating a separate silane primer step.
Acryloxy Silanes: Higher Reactivity, Lower Durability — a Tradeoff Formulators Cannot Ignore
Acryloxy silanes have a free-radical propagation rate constant (kp) roughly an order of magnitude higher than methacrylate analogs under equivalent conditions.True
Acrylate monomers consistently show kp values in the range of 15,000–30,000 L/mol·s at 25°C compared to 400–1,000 L/mol·s for methacrylates, a well-established kinetic difference documented in polymer chemistry literature.
That higher reactivity accelerates cure and improves conversion at low initiator doses — useful in high-throughput UV-line applications. But the unsubstituted ester in acryloxy silanes hydrolyzes faster under acidic or alkaline service conditions than the methacrylate version. In outdoor composite or marine applications, this durability gap becomes a real degradation pathway: the ester linkage breaks before the siloxane bond fails, leaving the organic matrix decoupled from the filler surface even while the silane anchor to the substrate remains intact. Choosing between the two requires knowing your service environment’s pH, moisture exposure, and temperature profile — not just your cure-speed target.
An operational warning applies here: vinylsilanes and methacryloxy silanes are often stored together in distributor warehouses, but their shelf-stability profiles differ. VTMS is sensitive to moisture ingress causing premature hydrolysis and self-condensation; MPS is more vulnerable to thermal polymerization of the methacrylate group above roughly 40 °C without inhibitor. Receiving inspections should check both water content (by Karl Fischer, target below 200–500 ppm depending on application) and inhibitor level — two separate failure modes requiring two separate tests.
Mercapto and Sulfide Functional Groups: Thiol Chemistry and the Sulfur Bridge in Tire Technology
Sulfur-containing silanes occupy a narrower commercial niche than amino or epoxy types, but within that niche—primarily silica-reinforced rubber and surface functionalization of coinage metals—they are irreplaceable. Two structurally distinct subfamilies dominate: monofunctional mercaptosilanes carrying a single thiol terminus, and the bis-silyl polysulfide TESPT, whose architecture is fundamentally different from every other silane coupling agent class.
3-Mercaptopropyltrimethoxysilane (MPTMS): Thiol Reactivity at the Molecular Level
MPTMS carries the structure (MeO)₃Si–C₃H₆–SH. The thiol group (–SH) is a soft nucleophile in the Pearson HSAB sense, which gives it high affinity for soft Lewis acid surfaces—gold, copper, silver—where harder oxygen-based nucleophiles bond far less efficiently. This makes MPTMS the standard choice for forming self-assembled monolayers on gold electrodes in biosensor fabrication and for passivating copper traces in printed circuit board laminates.
The pKa of an aliphatic thiol sits around 10–11. At neutral pH, well below that pKa, the –SH form dominates rather than the thiolate anion (–S⁻). That matters practically: thiol–ene click chemistry, where –SH adds across a C=C double bond under UV or thermal initiation, proceeds through a radical mechanism that requires the protonated thiol form. At elevated pH the thiolate anion is a better nucleophile but a worse radical precursor. Formulators using MPTMS as a latent epoxy hardener exploit the nucleophilic thiolate at mildly alkaline conditions; those running UV-cure coatings run the reaction at neutral pH to keep the thiol protonated.
One operational caution: thiol oxidation during storage converts –SH groups to disulfide dimers (–S–S–). Even partial oxidation reduces active coupling site density measurably. Drums should be blanketed with nitrogen and opened under inert conditions once the seal is broken.
TESPT (Si69): Structural Logic of the Polysulfide Bridge
Bis(triethoxysilylpropyl) tetrasulfide—universally called TESPT or Si69—has the formula (EtO)₃Si–C₃H₆–Sₓ–C₃H₆–Si(OEt)₃, where x averages 3.5–3.7 sulfur atoms but spans a distribution from x = 2 to x = 6. That distribution is not an impurity profile; it is an intrinsic feature of polysulfide synthesis. The mean sulfur rank depends on the reaction stoichiometry and temperature used in production, and suppliers typically specify an average value rather than a single integer.
TESPT contains exactly 4 sulfur atoms in every moleculeFalse
Commercial TESPT contains a distribution of polysulfide chain lengths (x = 2 to 6), with a mean around 3.5–3.7. Describing it as a pure tetrasulfide is a simplification that misleads formulation work.
The bifunctional architecture is what separates TESPT from every single-headed silane in this article series. Each molecule presents two independent triethoxysilyl groups. During silica compounding in a rubber mixer, each silyl head can hydrolyze and condense onto a separate silica particle surface. The result is a covalent bridge—silica particle → Si–O–silica bond → propyl spacer → polysulfide chain → propyl spacer → Si–O–silica bond → second silica particle—that both ties silica particles into the rubber network and provides the sulfur reservoir for vulcanization. No single-head silane creates this stitching geometry.
Processing Window and Bond Dissociation Energetics
TESPT is added during the non-productive mixing stage at 140–160°C. The chemistry at that temperature is selective by design. The S–S bond dissociation energy in a polysulfide falls in the range of roughly 150 kJ/mol, while the C–S bond along the propyl spacer is approximately 270 kJ/mol. At mixing temperatures the polysulfide bridge cleaves preferentially, releasing sulfur radicals and short-chain sulfide species—monosulfides and disulfides—that participate in rubber vulcanization. The propyl–silane portion and the Si–O–silica bonds, being substantially stronger, remain intact throughout mixing.
This selectivity is not accidental. A silane that released its silica bond at mixing temperature would destroy the filler coupling function before the tire compound ever reached the cure press. Formulating outside the 140–160°C window—running too hot—accelerates premature sulfur release and scorch; running too cool leaves the polysulfide bridge uncleaved and sulfur unavailable for crosslinking.
Blocked Mercaptosilanes: Odor Control Without Sacrificing Function
Unblocked mercaptosilanes have odor detection thresholds in the low ppb range. In a production environment processing hundreds of kilograms per batch, even trace vapor is an air-quality and worker-safety concern that can trigger regulatory attention. The engineering solution is thiocarbamate-blocked mercaptosilanes, where the –SH hydrogen is replaced with a –S–C(=O)–NR₂ protecting group. The blocked molecule has negligible odor under ambient storage and handling conditions. At rubber processing temperatures—typically above 150°C—the thiocarbamate group cleaves in situ, regenerating the free thiol exactly when and where it is needed.
The trade-off is a slightly narrower deprotection window and sensitivity to moisture during storage that can prematurely hydrolyze the blocking group. Procurement teams specifying blocked mercaptosilanes should confirm shelf-life certification under the actual warehouse humidity conditions at their plant, not just the supplier’s standard specification humidity.
Chloro, Isocyanato, and Specialty Functional Groups: Structures for High-Reactivity and Niche Applications
Amino, epoxy, and vinyl silanes dominate commercial volumes for good reason — they cover the majority of polymer composite and adhesion applications. But a procurement manager who only knows those three classes will eventually face a substrate, polymer matrix, or process condition where none of them work cleanly. Chlorosilanes, isocyanatosilanes, ureidosilanes, and aromatic-functional variants each exist because a specific chemical problem demanded a structural solution that the mainstream classes could not provide.
Chlorosilanes as Synthetic Intermediates, Not Direct Coupling Agents
Chloropropyltrimethoxysilane (ClC₃H₆Si(OCH₃)₃) looks superficially like an aminosilane with the nitrogen replaced by chlorine, but its industrial role is fundamentally different. The primary alkyl chloride at the organofunctional end is not a coupling group in the adhesion sense — it is a handle for post-functionalization chemistry. The C–Cl bond energy sits around 330–340 kJ/mol, weaker than C–F (around 485 kJ/mol) and selectively reactive toward SN2 displacement by hard nucleophiles: primary amines, thiols, azides, and organolithium species all react cleanly in polar aprotic solvents like DMF or DMSO. This makes chloropropylsilane the preferred starting material when a specialty organofunctional group cannot tolerate the conditions of direct silicon chemistry — you build the silane backbone first, then install the functional group via the chloride.
The operational catch is that the C–Cl bond is reactive enough to cause problems if storage conditions are poor. Trace moisture or protic solvents will generate HCl slowly, which then catalyzes premature hydrolysis of the methoxysilane end. Drums should stay sealed, dry, and away from ambient humidity. pH indicators in the headspace of storage tanks are a low-cost early-warning measure that many smaller compounders skip, to their eventual regret.

Isocyanatosilanes: Maximum Reactivity, Minimum Tolerance for Moisture
3-Isocyanatopropyltriethoxysilane (ICPTES) carries the –N=C=O group, and that cumulated double-bond system is among the most electrophilically reactive functional groups in practical silane chemistry. Second-order rate constants for reaction with primary alcohols at room temperature span roughly 10–1000 L/mol·s depending on the alcohol’s steric environment and whether a tin or amine catalyst is present — meaning at process concentrations, this reaction can be essentially instantaneous. That reactivity is exactly why isocyanatosilanes are used to couple inorganic surfaces directly into polyurethane systems: the silane hydrolyzes and bonds to the mineral surface on one end, while the isocyanate reacts with hydroxyl or amine groups in the polymer on the other, all without a separate coupling catalyst step.
Isocyanatosilanes can react with atmospheric moisture fast enough to cause significant functional group loss during open-vessel handling at typical plant-floor humidity levels.True
The –N=C=O group reacts with water to form a carbamic acid intermediate that decomposes to an amine and CO₂. At 50–60% relative humidity, measurable isocyanate consumption can occur within minutes of air exposure, which is the same moisture sensitivity seen in MDI and TDI handling in polyurethane production.
Dry storage is non-negotiable: nitrogen blanketing, moisture-barrier packaging, and cold-chain shipping in humid climates are standard requirements rather than premium options. A plant that treats ICPTES like a standard triethoxysilane will see inconsistent surface coverage, poor adhesion development, and batch-to-batch variability that is genuinely difficult to trace back to the root cause without systematic moisture auditing.
Ureidosilanes: Trading Reactivity for Durability
The ureido group (–NH–CO–NH₂) can be understood structurally as what happens when an isocyanatosilane reacts with water or when an aminosilane reacts with CO₂ — two urea nitrogens flanking a carbonyl. That symmetry is not accidental; it produces a functional group with considerably higher hydrolytic stability than the parent isocyanate and superior long-term wet adhesion compared to a simple aminosilane. In mineral-filled polyamide composites exposed to automotive coolant cycling — a service environment that cycles between roughly 90–130°C and involves prolonged aqueous glycol contact — ureidosilane-treated glass fiber retains interfacial bond integrity where aminosilane-treated fiber begins to show delamination within hundreds of hours of aging. The hydrogen-bonding capacity of both NH groups contributes to adhesion in polar matrices like polyamide and polyurethane without requiring the strong nucleophilicity that makes aminosilanes problematic in some catalyst-sensitive systems.
Aromatic and Styryl Variants: Pi-Stacking and Optical Applications
Styrylsilanes and phenylamino silanes introduce aromatic rings into the organofunctional architecture. The practical consequence is pi–pi stacking interactions with aromatic polymer matrices — polystyrene, ABS, polycarbonate — which provide interfacial compatibility that an aliphatic silane simply cannot replicate through dispersion forces alone. UV-absorbing capacity is a secondary benefit exploited in antireflective coatings and optical fiber buffer layers, where refractive index matching between the silane interlayer and the polymer matrix is as important as mechanical adhesion. These are low-volume, high-margin applications; the silane cost per unit area of treated surface is rarely the dominant factor in the bill of materials.
Bifunctional and Hybrid Architectures: Designer Molecules for Complex Composites
The structural frontier involves silanes carrying two chemically distinct Y groups tethered to the same silicon center on separate carbon chains. An amino-epoxy hybrid silane, for example, can sequentially react with an amine-reactive substrate surface through its epoxide while simultaneously serving as an epoxy-reactive component through its amine — effectively acting as a molecular bridge between two chemically incompatible interfaces. Commercial examples remain specialized, synthesis costs are substantially higher than single-functional grades, and qualifying them through a supply chain is a multi-quarter project. But in multilayer composite structures where a single-functional silane creates a weak boundary layer rather than a strong interphase, the hybrid architecture resolves the problem at the molecular level rather than through process workarounds.
Hydrolysis Stability, Shelf Life, and How Molecular Structure Governs Storage and Handling Requirements
Understanding the reactivity hierarchy of the X-group — the inorganic-facing hydrolyzable substituent — is not academic. It determines whether your bulk shipment arrives as a usable liquid or a partially condensed gel, and whether your application line can use standard nitrogen-purged drums or needs dedicated cold-chain logistics.
Hydrolysis Rate Hierarchy of the X Group
The order is unambiguous: chlorosilanes hydrolyze fastest, followed by acetoxysilanes, then methoxysilanes, then ethoxysilanes. Chlorosilanes exposed to atmospheric moisture at typical warehouse humidity (50–70% RH) begin reacting in seconds, releasing HCl and forming silanols and siloxane oligomers within minutes. That corrosive byproduct alone disqualifies chlorosilanes from most bulk storage scenarios. Acetoxysilanes are slower but still release acetic acid on hydrolysis — a practical concern for any substrate sensitive to pH drift.
Ethoxysilanes are the workhouse of bulk supply for exactly this reason. Their hydrolysis half-life under ambient humidity conditions can range from 12 months to well over 24 months, depending on container headspace moisture, storage temperature, and whether the product was filled under dry nitrogen. Methoxysilanes fall between ethoxysilanes and acetoxysilanes — roughly 3–8 times faster hydrolysis rate than their ethoxy counterparts at equivalent conditions, which matters when choosing between KH-550 analogs offered in both methoxy and ethoxy variants.
Functional Group Sensitivity at the Organofunctional End
The Y-group side has its own vulnerability ranking, independent of the X-group chemistry. Isocyanatosilanes are the most sensitive: the –NCO group reacts readily with atmospheric moisture and any protic nucleophile, so these products require nitrogen blanket storage, moisture-proof packaging, and cold-chain shipping in the 5–10°C range for any shipment exceeding a few weeks. Epoxy (glycidoxy) silanes are relatively stable under neutral to mildly alkaline conditions but undergo accelerated ring-opening in acidic environments — storing them in contact with moisture at pH below 4 progressively destroys the reactive oxirane. Mercaptosilanes are vulnerable to oxidation: the –SH group can form disulfide linkages during prolonged storage in oxygen-rich headspace, which is why reputable suppliers add low-level antioxidant stabilizers and specify nitrogen blanketing for long-term storage. Amino silanes are comparatively robust against hydrolysis at the organofunctional end, though they can absorb CO₂ from air to form carbamate salts that affect reactivity. Vinyl silanes sit at the stable end of this scale — the C=C double bond is not hydrolytically labile under normal storage conditions, making vinyltriethoxysilane among the most straightforward grades to warehouse.
| Functional Group | Primary Degradation Mechanism | Recommended Storage | Practical Shelf Life (sealed, dry) |
|---|---|---|---|
| Isocyanato (–NCO) | Moisture reaction, urethane/urea formation | N₂ blanket, 5–10°C, moisture-proof | 6–12 months |
| Epoxy/Glycidoxy | Ring-opening under acid or moisture | Cool, dry, neutral pH | 12–18 months |
| Mercapto (–SH) | Oxidation to disulfides | N₂ blanket, antioxidant-stabilized | 12–24 months |
| Amino (–NH₂, –NH–) | CO₂ absorption, carbamate formation | Sealed, dry, cool | 18–24 months |
| Vinyl (C=C) | Polymerization (high temp/light) | Away from heat/UV | 24+ months |
Concentration Effects in Aqueous Application Solutions
This is where formulators routinely create problems for themselves. Once a trialkoxysilane is hydrolyzed in water, the resulting silanol intermediate is thermodynamically unstable above roughly 2% concentration — condensation oligomers form and can gel irreversibly within a few hours at room temperature. Industrial application practice targets 0.5–1.0% active silane in water, adjusted to pH 4–5 where the hydrolyzed intermediate has maximum kinetic stability. Solutions should be prepared fresh or used within 4–8 hours of preparation unless the specific grade has been formulated with a stabilizing co-solvent.
Above 2% concentration, hydrolyzed trialkoxysilane solutions can gel irreversibly within hours at room temperature.True
Silanol condensation (Si–OH + HO–Si → Si–O–Si + H₂O) is concentration-dependent; exceeding ~2% pushes the equilibrium toward oligomeric and polymeric siloxane networks, which precipitate or gel and cannot be redissolved.
Temperature, Arrhenius Behavior, and Cold-Chain Logic
Every 10°C increase in storage temperature roughly doubles the hydrolysis rate for methoxysilanes — consistent with Arrhenius kinetics and an activation energy in the 60–80 kJ/mol range typical for this reaction class. A drum of methoxysilane stored at 35°C in an unventilated warehouse degrades at roughly four times the rate it would at 15°C. For sensitive grades — isocyanato and chloro variants especially — cold-chain logistics at 5–15°C are not a premium option but a technical requirement to preserve molecular integrity across shipping times that can reach 4–6 weeks for ocean freight to distant markets.
Quality Indicators That Reflect Molecular Integrity
The specification parameters on a certificate of analysis are a direct readout of how well molecular structure has been preserved. GC purity of ≥98% confirms that hydrolysis and condensation side reactions have been controlled during manufacturing and filling. Residual methanol or ethanol content — the alcohol byproduct released during hydrolysis — serves as a sensitive early-warning marker: even slight in-drum moisture ingress will elevate this value before appearance changes. Water content by Karl Fischer titration, with a target below 200 ppm for moisture-sensitive grades, is the most immediate indicator of packaging integrity. Refractive index rounds out the picture, offering a fast, instrument-simple check that correlates with both concentration and oligomer content.
SiliconChemicals reports all four parameters on product certificates and provides application datasheets specifying maximum working concentration and preparation protocol for each grade — because the gap between a correctly stored, freshly prepared silane solution and a degraded one is often the difference between 90% fiber-matrix adhesion retention and a failed composite layup.
Frequently Asked Questions About Silane Coupling Agent Chemistry and Functional Groups
Does the choice between trimethoxysilane and triethoxysilane actually change how the silane bonds to a surface?
Both hydrolyze to the same silanol intermediate (≡Si–OH) and form identical Si–O–substrate covalent bonds once condensation is complete. The difference is kinetic and practical, not fundamental to the final bond. Trimethoxysilanes hydrolyze roughly 3–5× faster than their triethoxy counterparts at comparable pH and temperature — the precise rate depends on pH (fastest near 4–5), temperature, and water activity in the formulation. That speed advantage matters in fast-cure production lines where open time is measured in seconds. The trade-off is methanol as the hydrolysis by-product. In a confined mixing room or a coating booth without adequate ventilation, methanol accumulation is a real VOC and toxicity concern, not a theoretical one.
Triethoxy variants hydrolyze more slowly, releasing ethanol — a meaningfully lower toxicity profile. They also offer better shelf life in moisture-sensitive storage because the ethoxy group is simply less reactive toward adventitious moisture. The organofunctional Y group on the opposite end of the molecule is entirely independent of this choice. An aminopropyl trimethoxysilane and aminopropyl triethoxysilane carry identical amine reactivity; you are selecting between them on hydrolysis kinetics, by-product toxicity, and shelf stability, not on how the amine behaves with an epoxy resin.
Trimethoxysilane and triethoxysilane coupling agents form chemically equivalent Si–O–substrate bonds after hydrolysis.True
Both X-groups hydrolyze to the same silanol (Si–OH) species regardless of whether the leaving group is methanol or ethanol. The final condensation product at the substrate surface is structurally identical.
Can one silane coupling agent serve both a polyurethane and an epoxy adhesive in a manufacturing operation?
Generally no — and this is one of the most common procurement mistakes. The Y group must be reactive with the specific polymer matrix. Vinyl and methacryloxy silanes are designed for radical polymerization environments and are essentially inert toward urethane or isocyanate chemistry; using them as a primer under a PU adhesive will give you little more than a physisorbed coating.
The genuine exception is the amino class. Primary aminosilanes such as APTES (3-aminopropyltriethoxysilane) or the diamine DAMO react with epoxide rings via nucleophilic ring-opening to form stable beta-hydroxyamine bonds, and they react with isocyanate groups to form urea linkages. That dual reactivity makes amino-functional silanes unusually versatile across both epoxy and polyurethane systems — a real operational advantage when a plant runs both adhesive chemistries on the same substrate line and wants to rationalize its silane inventory.
Why doesn’t increasing silane application concentration reliably improve adhesion?
Past a critical coverage threshold, more silane makes adhesion worse, not better. The surface hydroxyl density on amorphous silica sits in the range of roughly 4–8 Si–OH groups per nm², and that finite density caps the number of covalent Si–O–substrate bonds that can form. Excess silane condenses onto itself, producing loosely cross-linked physisorbed multilayers. These multilayers are mechanically weak — they behave as a boundary layer that cohesively fails under stress before the actual adhesive bond ever loads. Optimal treatment concentrations are typically those producing 1–3 covalently anchored molecular layers. The exact optimum depends on substrate surface area, silane molecular footprint, and application method, but the structural logic is universal.
What silane is correct for silica-reinforced sulfur-vulcanized natural rubber?
TESPT — bis(triethoxysilylpropyl) tetrasulfide — is the established industry standard. Its two triethoxysilylpropyl heads anchor to precipitated silica surface silanols during mixing, while the polysulfide bridge (–S₄– on average, with a distribution of sulfur chain lengths) participates directly in sulfur vulcanization chemistry, covalently coupling the silica network into the rubber matrix. Mixing temperature control matters: TESPT should be incorporated at 140–155°C to ensure adequate silanization without premature scorch.
Mercaptopropyltriethoxysilane couples faster and can deliver marginally higher reinforcement efficiency, but the free thiol is a potent scorch accelerant. Processing windows narrow considerably, and temperature excursions during dump or transfer become a real risk. Some compounders use blocked mercaptosilanes to recover the reactivity advantage while reducing scorch sensitivity.
How do you confirm that a silane has actually bonded to a substrate surface rather than just deposited on it?
Three techniques, used in combination, give a reliable answer. DRIFTS (diffuse reflectance infrared Fourier transform spectroscopy) is the most accessible: it detects Si–O–substrate stretching bands in the 1000–1100 cm⁻¹ region and organofunctional group absorptions — N–H stretch near 3300 cm⁻¹ for amino silanes, C=O near 1720 cm⁻¹ for methacryloxy types. The presence of these bands after thorough solvent rinsing is strong evidence of covalent attachment rather than loose physisorption.
XPS (X-ray photoelectron spectroscopy) provides elemental surface composition and, critically, binding energy shifts that distinguish covalently bound silicon from physisorbed silane oligomers. For sulfur-containing silanes, XPS sulfur quantification confirms actual surface coverage.
Water contact angle measurement is the fastest shop-floor-compatible check. Untreated amorphous silica typically gives an advancing contact angle below 30°. A successful hydrophobic silane treatment pushes that above 70°. It won’t tell you the exact bonding mechanism, but a consistent contact angle result across a treated batch confirms process uniformity in minutes rather than hours.














